Publication in Science of Zhou Qilin and Zhu Shoufei's Group: Highly enantioselective carbene insertion into N–H bonds of aliphatic amines
On November 22, 2019, Zhou Qilin and Zhu Shoufei's team, Nankai University published an article entitled Highly enantioselective carbene insertion into N–H bonds of aliphatic amines online in Science, which reported highly enantioselective carbene insertion into N–H bonds of aliphatic amines using two catalysts in tandem. This research not only solves the long-term challenges of enantioselective carbene insertion reactions, but also provides a potential general strategy for transition metal-catalyzed asymmetric transformations involving strong coordination substrates. Eric N. Jacobsen from Harvard published a review article entitled A catalytic one-two punch in the Science with the same issue number, which systematically interpreted the research and gave it a high evaluation.
First Author: Li Maolin
Corresponding author: Zhou Qilin
Communication unit: Nankai University
Aliphatic amines strongly coordinate, and therefore easily inhibit, the activity of transition-metal catalysts, posing a marked challenge to nitrogen-hydrogen (N–H) insertion reactions. Here, we report highly enantioselective carbene insertion into N–H bonds of aliphatic amines using two catalysts in tandem: an achiral copper complex and chiral amino-thiourea. Coordination by a homoscorpionate ligand protects the copper center that activates the carbene precursor. The chiral amino-thiourea catalyst then promotes enantioselective proton transfer to generate the stereocenter of the insertion product. This reaction couples a wide variety of diazo esters and amines to produce chiral α-alkyl α–amino acid derivatives.
Chiral amines are ubiquitous in natural products, pharmaceuticals, and agrochemicals. Approximately 43% of the top 200 prescription medicines in 2016 contain an aliphatic amine moiety (Fig. 1A). The development of highly enantioselective transition-metal–catalyzed reactions that form C–N bonds is thus of long-standing interest in synthetic chemistry. Transition-metal–catalyzed carbenoid insertion into N–H bonds has proven a straightforward method in this respect, benefitting from mild reaction conditions, good functional group tolerance, and readily available reactants. Recently, chiral transition-metal catalysts have been successfully applied to enantioselective N–H insertion reactions in the synthesis of natural or unnatural chiral α–amino acid derivatives. However, these reactions have been restricted to aromatic amines or amides (Fig. 1B). Aliphatic amines are comparatively stronger Lewis bases and thus poison the metal catalysts by strong coordination, interfering with generation of the metal carbenoid. Moreover, excess aliphatic amines can displace the ylide from metal-ylide intermediates, leading to racemic product formation from the free ylide (Fig. 1C, upper).
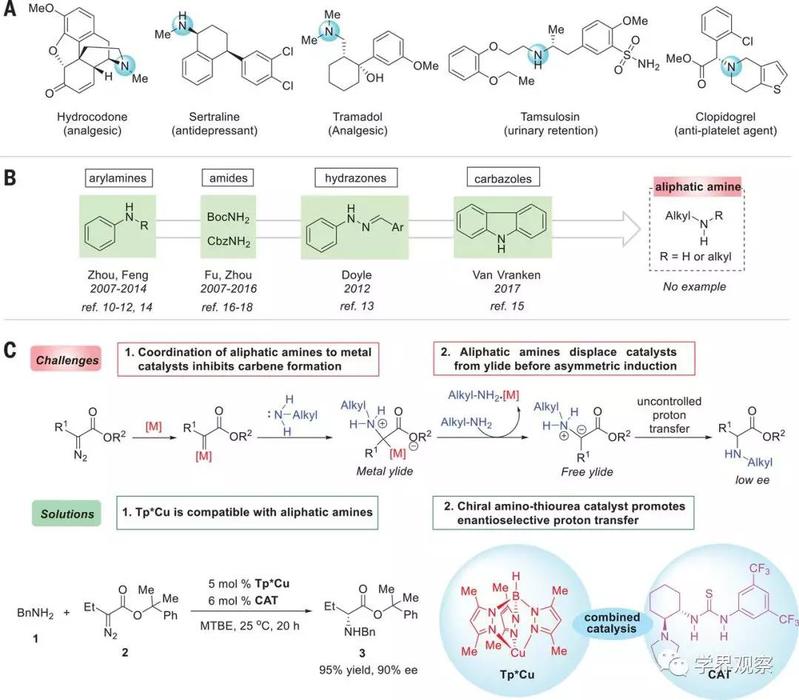
Fig. 1 ******** for enantiocontrol of N–H insertion reactions of aliphatic amines with carbenes.
(A) Representative drugs demonstrating the ubiquity of chiral aliphatic amines in bioactive molecules. (B) Amine sources reported for enantioselective N–H insertion reactions. (C) Enantioselective transition-metal–catalyzed N–H insertion reactions with aliphatic amines: challenges and solutions. Optimal reaction conditions: The reaction of 1 (0.2 mmol), 2 (0.22 mmol), Tp*Cu (5 mole %), and CAT (6 mol %) was carried out in 3 ml of methyl tert-butyl ether (MTBE) at 25°C for 20 hours. BnNH2, benzylamine; BocNH2, tert-butyl carbamate; CbzNH2, benzyl carbamate; Me, methyl; Et, ethyl; Ph, phenyl; M, metal; ref., reference.
Academician Zhou Qilin and Professor Zhu Shoufei from Nankai University envisioned that a combination of two catalysts might address these challenges: An achiral transition-metal catalyst compatible with aliphatic amines would generate the ylide intermediate, and a separate chiral catalyst would then promote enantioselective proton transfer. After exploring various transition-metal catalysts and chiral H-bonding catalysts in the N–H insertion reaction of α-diazobutanoates with benzylamine (tables S1 to S5), the authors report here the success of this approach, pairing the homoscorpionate-coordinated copper complex Tp*Cu [Tp* is hydrotris(3,5-dimethylpyrazolyl)borate] with a chiral amino-thiourea (CAT) bearing a pyrrolidine motif (Fig. 1C, lower). The reaction provides efficient, highly enantioselective access to chiral α-alkyl α–amino acid derivatives bearing secondary and tertiary amino substituents, which are difficult to prepare by other methods.

Fig. 2 Scope of aliphatic amines and α-diazo esters in the enantioselective N–H insertion reaction.
Reaction conditions: amines (0.2 mmol), α-diazo esters (0.22 mmol), Tp*Cu (5 mol %), CAT (6 mol %), 3 ml MTBE, 25°C, 20 hours. Isolated yields are given. The ee values were determined by high-performance liquid chromatography. (A) Scope of aliphatic amines. (B) Application to enantioselective late-stage functionalization of pharmaceuticals. (C) Scope of α-diazo esters. *Diazo esters (0.3 mmol), 36 hours. †Diazo esters (0.3 mmol), MTBE:CH2Cl2 = 10:1, 36 hours. ‡Diazo esters (0.3 mmol), 40°C, 20 hours. tBu, tert-butyl; iPr, iso-propyl.
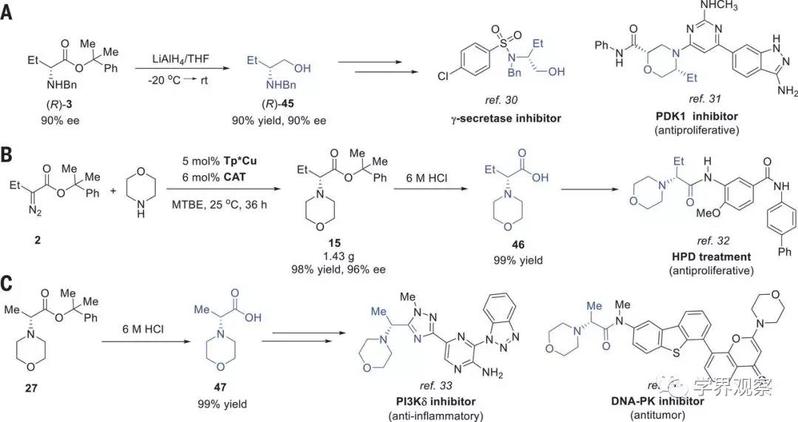
Fig. 3 Synthetic transformations of the N−H insertion products.
(A) Transformation of 3 to (R)-2-benzylamino-butanol [(R)-45], a key intermediate for the synthesis of bioactive molecules. THF, tetrahydrofuran; rt, room temperature. (B) Formal synthesis of HPD treatment agents with the N−H insertion as key step. (C) Transformation of 27 to 47, a key intermediate for the synthesis of bioactive molecules.
To gain a deeper understanding of the mechanism of the NH insertion reaction, the authors performed kinetic analysis using online infrared (IR) spectroscopy. In a nutshell, the kinetic, NMR, and UV studies are consistent with the Tp*Cu•CAT complex, rather than the Tp*Cu•BnNH2 complex, as the resting state of the catalyst in the reaction. Although the Tp*Cu•CAT complex is the main resting state of the copper, free Tp*Cu is still evident under the reaction conditions (fig. S10) and can react with the diazo compound.
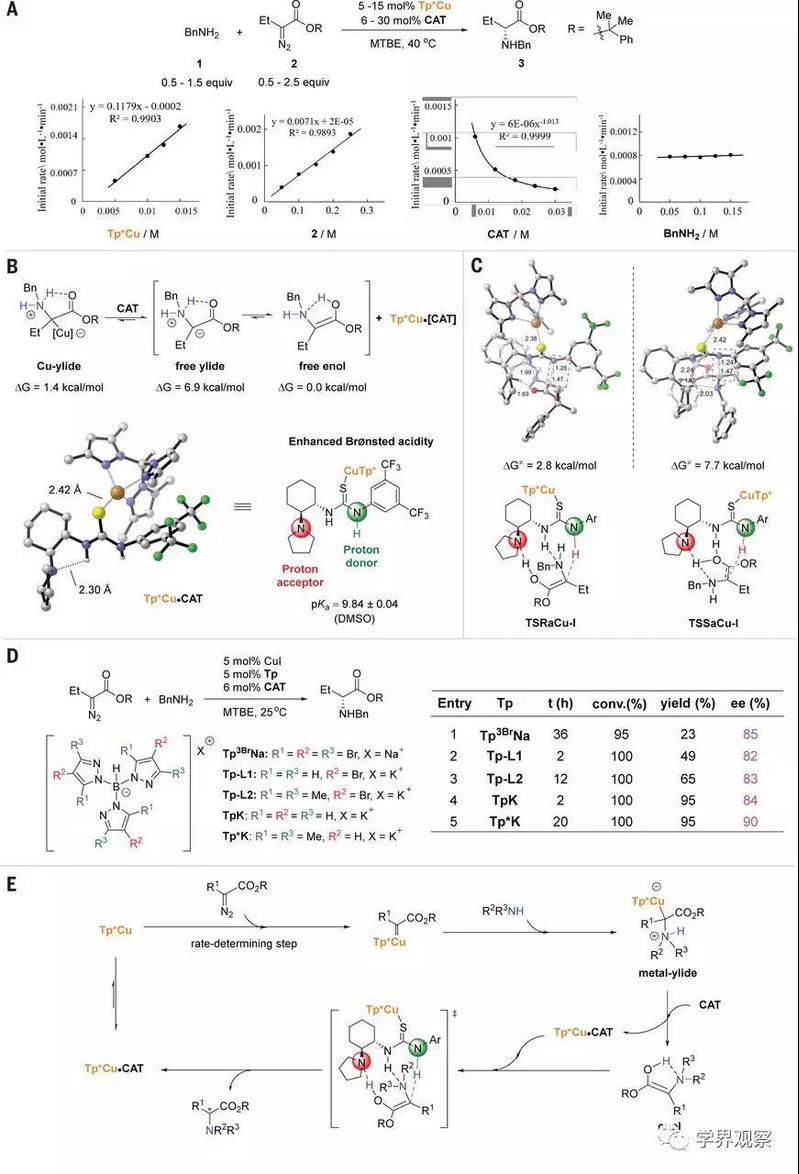
Fig. 4 Mechanistic studies.
(A) Kinetic profiles of Cu-catalyzed N−H insertion reaction of 2 and BnNH2. (B) Calculated Gibbs free energy (ΔG) of Cu-ylide, free ylide, and free enol. Lowest-energy ground-state structure of the Tp*Cu•CAT complex. Structures of alternative higher-energy complexes are provided in fig. S20. (C) DFT-optimized lowest-energy transition structures for R and S products. Calculations were performed at the m062x-D3/def2tzvpp//m062x-D3/def2svp level. Structures of alternative higher-energy complexes are provided in figs. S21 to S25. (D) Influence of different Tp ligands. (E) Proposed catalytic cycle for the enantioselective carbene insertion into N–H bonds of aliphatic amines. conv., conversion; DMSO, dimethyl sulfoxide; equiv, equivalents.
Several other tris(pyrazolyl)borate (Tp) ligands bearing different substituents on the pyrazol rings were also evaluated under the standard reaction conditions (Fig. 4D). Despite a large fluctuation in the yield, the in situ IR studies showed that all tested Tp ligands promoted high conversions and that the major by-product was 2-phenylpropan-2-yl but-2-enoate, resulting from the β-H migration of the metal carbenoid (figs. S15 to S19). Modifying the Tp ligands also influenced the enantioselectivity when the same chiral thiourea catalyst was used, indicating involvement of the copper catalyst in the enantio-determining step. By contrast, upon tuning the electronic properties of the arene ring of the chiral thiourea catalyst, the enantioselectivity decreased precipitously, whereas the yield remained almost unchanged (table S7). We again hypothesize that copper coordination enhances the Brønsted acidity of the thiourea catalyst while minimally influencing the distant site of enantioinduction (Fig. 4C).
On the basis of the aforementioned mechanistic studies, a catalytic cycle is proposed in Fig. 4E. The Tp*Cu•CAT complex serves as a resting state of the catalyst and dissociates to release Tp*Cu, which catalyzes transformation of the diazo ester into the metal carbenoid in the rate-determining step. Nucleophilic attack on the metal carbenoid by the aliphatic amine generates a metal ylide. The catalyst CAT displaces the ylide from the metal-ylide intermediate to generate free enol and the Tp*Cu•CAT complex. The Tp*Cu•CAT complex then promotes proton transfer in the free enol through a push-pull mechanism: The amino moiety accepts a proton from the hydroxy group of the enol while the thiourea moiety donates a proton to the β-carbon of the enol.
The author states that the success of the overall conversion depends on the comprehensive performance of achiral copper catalysts and chiral organic catalysts. This study not only addresses the long-term challenges of enantioselective carbene insertion reactions, but also provides a potential general strategy for transition metal-catalyzed asymmetric transformations involving strong coordination substrates.
The author states that the success of the overall transformation relies on the combined properties of the achiral copper catalyst and chiral organocatalyst. This study not only solves a long-standing challenge in enantioselective carbene insertion reactions but also provides a potentially general strategy for transition-metal–catalyzed asymmetric transformations involving strongly coordinating substrates.
Harvard University Jacobsen commented on the research during the same period of Science, stating that The cooperative action of achiral transition metal complexes with chiral hydrogen-bond donors holds enormous potential for achieving new asymmetric transformations. Organotransition-metal chemistry provides access to a wealth of reactivity modes inaccessible to organocatalysts, and chiral hydrogen-bond-donor catalysts have been found to promote enantiocontrol through a rich variety of noncovalent mechanisms. The system developed by Li et al. represents a compelling glimpse into some of the possibilities.

Academician Zhou Qilin, organic chemist, professor of Nankai University. Born in Nanjing, Jiangsu in February 1957. He graduated from the Department of Chemistry of Lanzhou University in July 1982, and obtained his master's and doctoral degrees from Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 1985 and 1987, respectively. He has performed postdoctoral research at Max-Planck Polymer Research Institute in Germany, Basel University in Switzerland, and Trinity University in the United States. In 2009 he was elected an academician of the Chinese Academy of Sciences. Mainly engaged in asymmetric catalytic synthesis. It involves the structural design of chiral catalysts, catalyst synthesis, corresponding asymmetric synthesis reactions and the application of these chiral catalysts to the synthesis of chiral molecules. A new class of chiral spiro ligands was developed, and a series of new chiral spiro catalysts were designed and synthesized based on this kind of ligands. These catalysts show excellent catalytic activity and enantioselectivity in a series of asymmetric synthesis reactions such as asymmetric catalytic hydrogenation, carbon-carbon bond and carbon-heteroatom bond formation. The research results have been applied in the synthesis of hands and things.

Professor Zhu Shoufei, received a Bachelor of Science degree and a Doctor of Science degree from the School of Chemistry, Nankai University in 2000 and 2005, respectively, and a postdoctoral fellow at the University of Tokyo, Japan from 2012 to 2013. He has worked in the School of Chemistry, Nankai University from 2005 to present, and received the National Natural Science Fund in 2016 Funded by the Outstanding Youth Fund. He has long been engaged in the research of catalytic organic synthetic chemistry, focusing on several types of important organic synthesis reactions that take hydrogen transfer as the key step, and proposed the concept of chiral proton shuttle, which provides a new solution for metal-catalyzed asymmetric proton transfer reactions. ; Discovered the catalytic carbene insertion reaction of boron-hydrogen bonds, provided a new method for the synthesis of organoboron compounds; developed a variety of efficient catalysts for chlorination and hydrosilylation of olefins, and realized a variety of important biologically active molecules Efficient synthesis. He has published more than 100 research papers and authored two chapters: 5 Chinese patents have been authorized. He has won 2 first prizes in Tianjin Natural Science (both 3rd person), Youth Chemistry Award of Chinese Chemical Society, Youth Chiral Chemistry Award of Chinese Chemical Society, Tianjin Youth Science and Technology Award, Tianjin Youth May Fourth Medal, Asia Core Program Lectureship Award and other awards.

First author Mao-Lin Li of Nankai University
References:
https://science.sciencemag.org/content/366/6468/990
https://science.sciencemag.org/content/366/6468/948
Address: 94 Weijin Road, Tianjin 300071, CHINA
TEL: +86-22-2350 8470 E-mail: nkchem@nankai.edu.cn